

24 Dec 2025, JACS Au
Hang Qu, Fei Wang, Zhi-Chao Lei, Xin-Chang Wang, Liulin Yang, Xiao-Yu Cao, Chunhai Fan, and Zhong-Qun Tian*
Molecular assembly is a fundamental organizational principle in both living organisms and the fabrication of functional materials. However, artificial self-assembly systems lag far behind biological systems in terms of efficiency, controllability, complexity, and functionality. Here, inspired by catalysis in chemical reactions, we propose a novel strategy, termed as molecular catassembly, that employs catassemblers to dynamically manipulate cooperative multisite noncovalent interactions, thereby directing the pathway and accelerating assembly processes. By translating catalytic and biological principles into the catassembly, we summarize the distinctive features and multifaceted roles of catassemblers in manipulating cooperative multisite noncovalent interactions, facilitating mass transfer in crowded environments, and mediating energy transduction and feedback that endow systems with information-processing capabilities. Furthermore, we emphasize the pivotal role of catassemblers in multistep reaction-assembly cascades for the fabrication of hierarchical functional materials and the regulation of the cellular signaling pathway. We further elucidate how the integration of artificial intelligence technologies offers transformative potential to redefine the research paradigm of molecular (cat-)assembly. Nevertheless, the research of catassembly remains in its infancy and demands the integration of advanced concepts and methodologies from multiple disciplines. Such interdisciplinary efforts will be crucial for unraveling the complexity and functionality of molecular assembly, ultimately offering new perspectives and methodologies for both life sciences and soft matter research.

23 Dec 2025, Journal of the American Chemical Society
Zhihao Li, Xuehai Huang, Yansong Jiang, Wei Zeng, Ding Zou, Xue Dong, Liulin Yang, Xiaoyu Cao*, Zhongqun Tian*, and Yu Wang*
Chiral molecular assembly represents a frontier in creating life-inspired functional materials; yet, a predictive framework for its enantioselective catalysis remains elusive. Here, we introduce the Catassembly Triad – a mechanistic paradigm for catalytic enantioselective assembly, demonstrated using chiral supramolecular cages as a model system. Combining in situ characterization, ab initio calculations, and machine learning, we resolve how catalysts must harmonize attachment, chiral control, and detachment to direct assembly pathways. Kinetic and structural analyses reveal that chiral diamine catalysts (e.g., cyclohexanediamine) enforce enantioselectivity by templating transient intermediates with deformation-induced affinity changes driving timely catalyst release. We establish a predictive framework based on quantitative triad metrics (ΔEatt, ΔEctl, and ΔEdet) from calculations, validated by Bayesian clustering and experimental testing. The framework achieves a perfect concordance between predicted and observed catalyst efficacy across diverse molecular classes. This framework bridges enzymatic cooperativity and supramolecular assembly, enabling programmable chirality transfer and unlocking catalyst design for adaptive chiral materials.
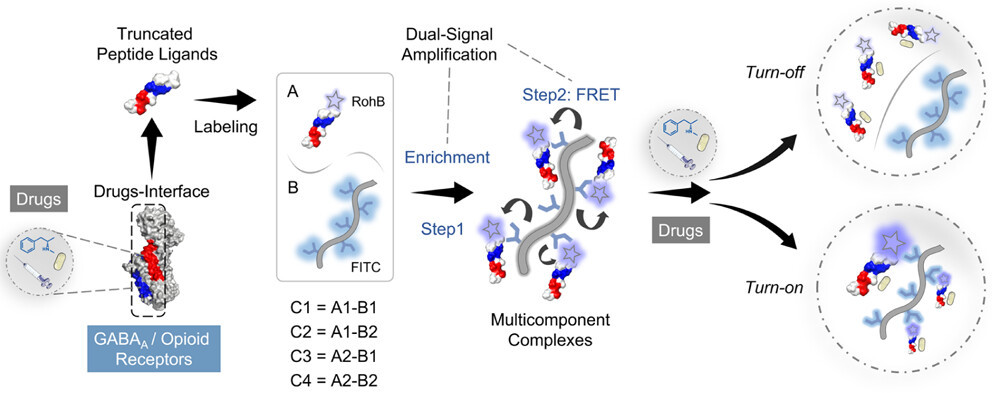
8 Dec 2025, ACS Analytical Chemistry
Shiqi Liuye, Tianhao Shi, Yang Zhou, Yuqing Chen, Yang Yu*, Liulin Yang*, and Xiaoyu Cao*
Synthetic illicit drugs have posed severe threats to health and social security. Differential sensing integrated with multivariate data processing methods provides substantial benefits for the simultaneous detection of multiple drugs. Rational design of sensing elements targets a variety of drugs, and achieving high sensitivity using nontargeted sensing elements remains a challenge. To address these challenges, we design peptide-based sensing elements through rational truncation of key recognition fragments from receptor proteins and propose a dual signal amplification strategy that leverages Förster resonance energy transfer (FRET) enhancement and macromolecule-induced probe enrichment. Two fluorogenic peptides are truncated from the binding domain of GABAA and opioid receptors. The peptides are then enriched by fluorescently tagged carboxymethyl dextran to form multicomponent complexes and trigger FRET signals. A three-channel four-complex sensor array is constructed, which can simultaneously identify 5 drugs (C = 100 nM) with 100% accuracy and different mixing ratios and achieve quantitative detection of drugs. Moreover, the array excels in distinguishing six simulated drug samples in the artificial urine. The sensor array constructed using this strategy combines high sensitivity with parallel detection capability in complex environments, offering a promising solution to the growing challenge of the detection of increasingly prevalent drugs.

5 Dec 2025, ACS Organic Letters
Mingyu Cui, Xue Dong, Guoqiang Jiang, Wei Zeng, Hang Qu, Andrew C.-H. Sue,* and Xiaoyu Cao*
A serendipitous outcome in the mandelic acid-mediated imine condensation of a tetraphenylethene-derived tetraaldehyde and tris(2-aminoethyl)amine led to [2 + 4] face-rotating molecular boxes, rather than the expected [6 + 8] cubic cages. The resulting D2-symmetric assemblies feature two homochiral tetraphenylethene facial units and four unreacted amine handles. Mandelic acid engages in cooperative hydrogen bonding, steric effect and C–H···π interactions that steer the assembly pathway and supramolecular chirality. NMR, ECD, and X-ray analyses confirm the formation of a pair of diastereomeric supramolecular complexes exhibiting distinct optical activity in solution.

14 Oct 2025, Scientific Data
Da Long, Zhihao Li, Xin Xu, Chengchun Liu, Hui Zhang, Xiaoyu Cao*, Liulin Yang*, Xinchang Wang* and Fanyang Mo*
Determining the absolute configuration of chiral molecules is of fundamental importance in the fields of natural products chemistry, asymmetric catalysis, and pharmaceutical development. A widely adopted approach involves the comparison of experimental and theoretical electronic circular dichroism (ECD) spectra, which has proven to be a reliable method for absolute configuration assignment. However, the generation of theoretical ECD spectra via time-dependent density functional theory (TD-DFT) remains the rate-limiting step in this workflow, making its acceleration both essential and challenging. Although recent advances in deep learning offer promising strategies for establishing structure-spectrum relationships and expediting theoretical spectrum prediction, the lack of standardized and comprehensive ECD spectral datasets continues to hinder progress. This study presents the Chiral Molecular Circular Dichroism Spectral (CMCDS) dataset, a systematically structural benchmark dataset that addresses the fragmentation of existing ECD data. Characterized by high standardization, scalability, and broad molecular diversity, CMCDS facilitates deep learning applications in ECD analysis and fosters data-driven discovery of chiral molecules.

1 Aug 2025, Crystal Growth & Design
Ding Zou*, Wenbin Gao, Tianyi Tong, Zhihao Li, Hanxun Zou, Liulin Yang*, and Xiaoyu Cao*
The rational design of chiral fluorescent organic molecular cages requires a comprehensive understanding of structure–activity relationships. In this study, we constructed two molecular face-rotating polyhedra (FRP), namely, FRT-3 and FRT-4, from flexible tris(2-aminoethyl)amine (TREN), rigid Tri-NH2, and tris(4-phenyl)aniline (TFPA). FRT-3 crystallized as a pair of enantiomers (homodirectional, 4M and 4P), while FRT-4 formed three diastereomers (heterodirectional: 3M1P, 3P1M, and 2M2P). To elucidate the effect of vertex geometry on photophysical behavior, we investigated their fluorescence properties in solvents of varying polarities and under protonation. Both FRT-3 and FRT-4 exhibit pronounced intramolecular charge transfer (ICT) behavior and show distinct responses to acids in solution. These findings offer valuable insights into the potential of chiral fluorescent molecular cages for sensing and optoelectronic applications.

18 Feb 2025, Chemical Science
Peichen Shi, Ganyu Chen, Qiang Chen, Huiting Wu, Suixu Li, XiaoYu Cao*, Liulin Yang*, and Zhong-Qun Tian*
The complexity of multi-component molecular assembly demands precise control strategies to enhance both efficiency and selectivity. Heterogeneous nucleation and the autocatalytic secondary pathway, as key regulatory strategies, have attracted widespread attention for their crucial roles in crystal growth and amyloid protein aggregation. Here, we apply a heterogeneous nucleation strategy to supramolecular polymer systems and report the first direct observation of surface-enrichment-induced primary nucleation and a spontaneous fragmentation-driven autocatalytic secondary process. A heterogeneous nucleating agent promotes primary nucleation, facilitating supramolecular chiral induction. The resulting chiral polymers undergo a catalytic cycle of fragmentation and re-growth at their termini, with the fragments also acting as seeds for nucleation and growth. These pathways play a crucial role in the polymerization process and are essential for chiral transfer and asymmetry amplification, enabling the achievement of maximum enantioselectivity with as little as 0.5% molar equivalent of the heterogeneous nucleating agent. Furthermore, we reveal the existence of an optimal equivalent in their catalytic kinetics, arising from a surface assembly mechanism. In this mechanism, monomers adsorbed on the surface of the heterogeneous nucleating agent assemble with those in solution, rather than through surface diffusion and assembly. This process resembles the surface-catalyzed Eley–Rideal mechanism. Our study highlights the potential of heterogeneous nucleation as an effective strategy for controlling supramolecular polymerization and offers new insights into its underlying mechanism.

9 Jan 2025, ACS Nano
Wang Li#, Yang Zhou#, Tianyi Tong, Sheng He, Congsen Wang, Xinran Zhang, XiaoYu Cao, Liulin Yang*, and Zhong-Qun Tian
The assembly of peptides is generally mediated by liquid–liquid phase separation, which enables control over assembly kinetics, final structure, and functions of peptide-based supramolecular materials. Modulating phase separation can alter the assembly kinetics of peptides by changing solvents or introducing external fields. Herein, we demonstrate that the assembly of peptides can be effectively catalyzed by complex coacervates. The negatively charged sodium alginate (SA) can form complex coacervates with the positively charged KLVFFAE (Aβ16–22, abbreviated as KE) peptide, thereby lowering the nucleation barrier and promoting the assembly of the peptide. As the binding affinity of SA-KE and the dosage of SA decrease, the system shifts from a relatively inefficient template-induced assembly to a highly efficient catalytic assembly before ultimately reverting to slow spontaneous assembly. Therefore, both the affinity as well as the stoichiometry do not follow the intuitive rule that “more is better”, but rather there exists an optimal value that maximizes the rate of assembly.







