
2022
18 Nov 2022, SCIENTIA SINICA Chimica
Hang Qu, Tianyi Tong, Zhi-Chao Lei, Peichen Shi, Liulin Yang, Xiaoyu Cao, Yiqin Gao, Zhonghuai Hou, Xin Xu and Zhong-Qun Tian*
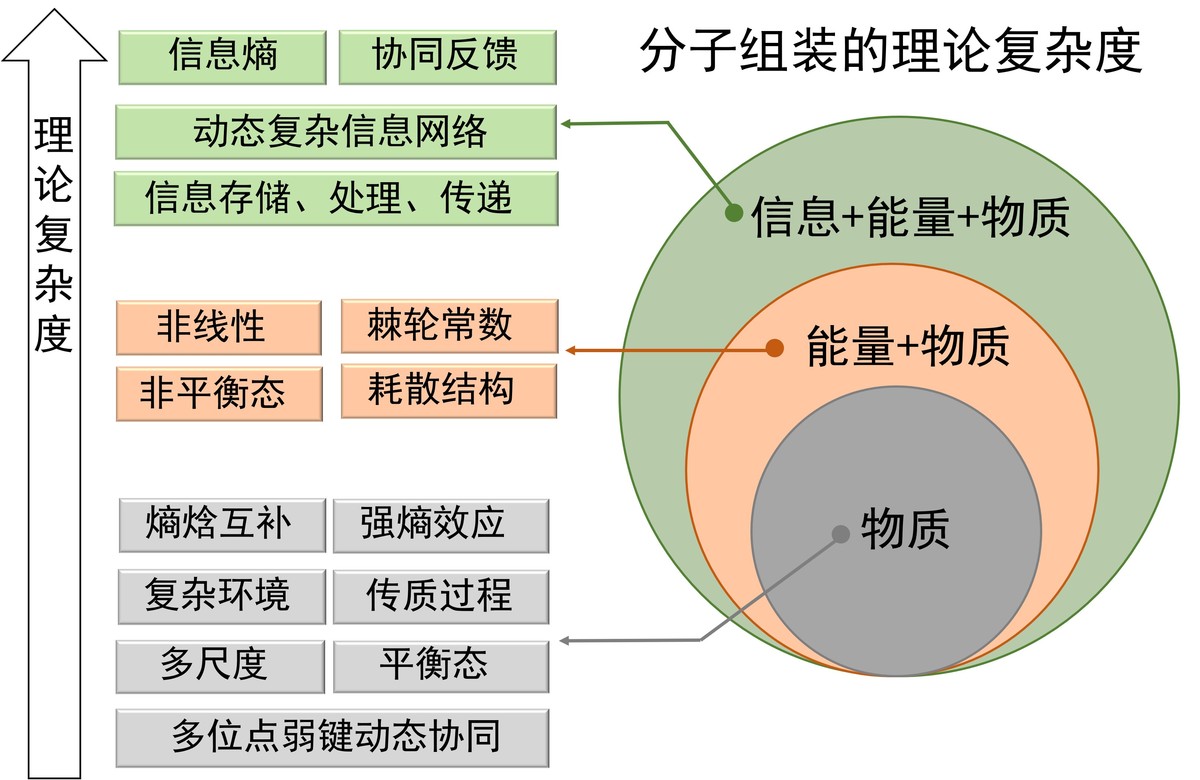
Molecular assembly (MA) is an essential approach to create novel substances and new functional materials beyond molecules. Improving the controllability of MA processes and the functionality of assemblies is the core
target in this field. However, due to the limitation of methods and theories for complex MA systems, most of current researches are based on the “black box” paradigm, which can mostly focus on the initial and final state of MA. Such a paradigm has become a bottleneck that restricts the development of MA. Here, we review the current theoretical methods and models of MA systems from the molecular to subcellcular level. We try to measure the complexity of MA research from the dimension of matter, energy and information, and gradually explore the current status, opportunities and breakthrough of the theoretical research of MA: in the dimension of matter, we illuminate the pathways of MA processes are multi-step, multi-pathway and multi-scale; in the integrated dimension of energy and matter, we demonstrate MA systems are usually accompanied by the entropy-driven phenomena or the complementation of entropy and enthalpy. We further discuss how the MA systems far from the equilibrium can form an order spatiotemporal dissipated structure; in the integrated dimension of information, energy and matter, we demonstrate how the synergetic processes of positive or negative feedback facilitates the emergence of complex physiological functions of MA systems. To address these challenges in the development of MA theoretical researches, we probably need to establish a larger theoretical framework and synergistically develop the research methods of MA from the three dimensions of matter, energy and information. It seems to help us to explore its fundamental laws of MA comprehensively, then establish the new theory and promote the development of assisting methods with high efficiency and, and finally elevate the complexity and functionality of MA system, which may provide new perspectives and methods for life science and soft matter science.
15 Nov 2022, Chemical Science
Shilin Zhang, Yulian Zhang, Huiting Wu, Zhihao Li, Peichen Shi, Hang Qu, Yibin Sun, Xinchang Wang, Xiao-Yu Cao, Liulin Yang* and Zhong-Qun Tian
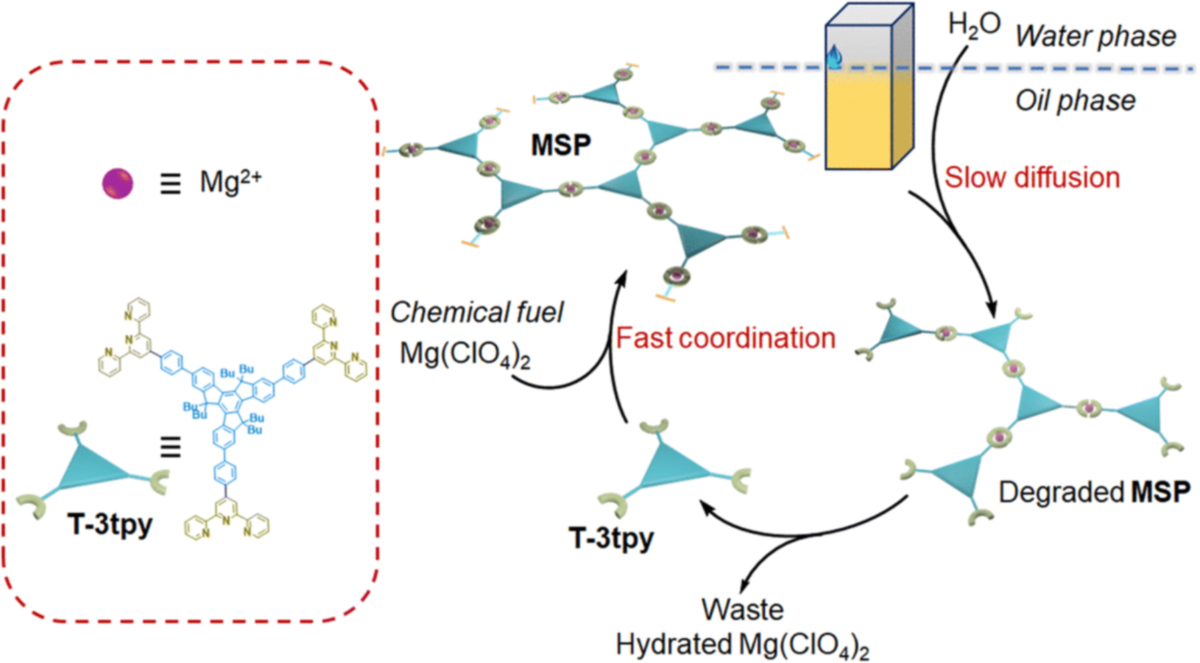
Inspired by life assembly systems, the construction of transient assembly systems with spatiotemporal control is crucial for developing intelligent materials. A widely adopted strategy is to couple the self-assembly with chemical reaction networks. However, orchestrating the kinetics of multiple reactions and assembly/disassembly processes without crosstalk in homogeneous solutions is not an easy task. To address this challenge, we propose a generic strategy by separating components into different phases, therefore, the evolution process of the system could be easily regulated by controlling the transport of components through different phases. Interference of multiple components that are troublesome in homogeneous systems could be diminished. Meanwhile, limited experimental parameters are involved in tuning the mass transfer instead of the complex kinetic matching and harsh reaction selectivity requirements. As a proof of concept, a transient metallo-supramolecular polymer (MSP) with dynamic luminescent color was constructed in an oil–water biphasic system by controlling the diffusion of the deactivator (water molecules) from the water phase into the oil phase. The lifetime of transient MSP could be precisely regulated not only by the content of chemical fuel, but also factors that affect the efficiency of mass transfer in between phases, such as the volume of the water phase, the stirring rate, and the temperature. We believe this strategy can be further extended to multi-compartment systems with passive diffusion or active transport of components, towards life-like complex assembly systems.
12 Oct 2022, Journal of American Chemistry Society
Yulian Zhang,‡ Shilin Zhang,‡ Huiting Wu, Xue Dong, PeiChen Shi, Hang Qu, Yuqing Chen,Xiao-Yu Cao, Zhong-Qun Tian, Xiaolan Hu,* and Liulin Yang*
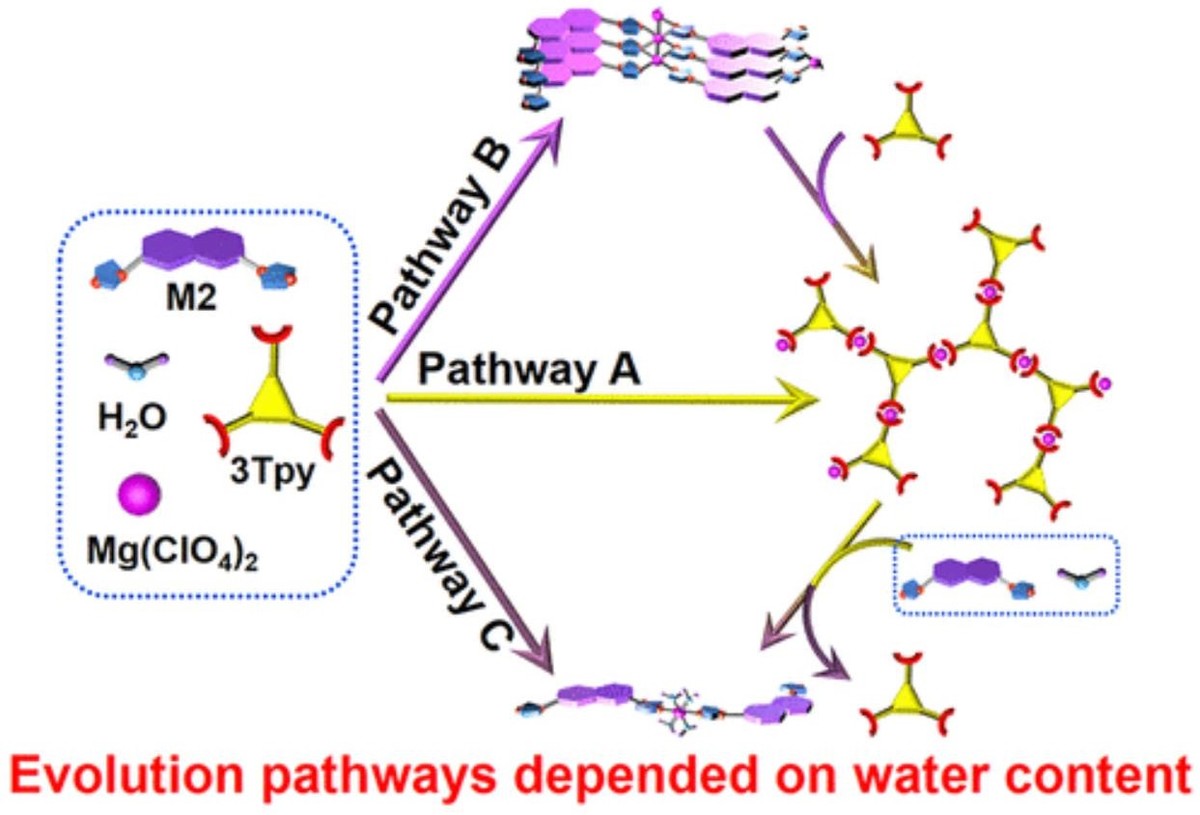
Trace water in organic solvents can play a crucial role in the construction of supramolecular assemblies, which has not gained enough attention until very recent years. Herein, we demonstrate that residual water in organic solvents plays a decisive role in the regulation of the evolution of assembled structures and their functionality. By adding Mg(ClO4)2 into a multi-component organic solution containing terpyridine-based ligand 3Tpy and monodentate imidazole-based ligand M2, the system underwent an unexpected kinetic evolution. Metallo-supramolecular polymers (MSP) formed first by the coordination of 3Tpy and Mg2+, but they subsequently decomposed due to the interference of M2, resulting in a transient MSP system. Further investigation revealed that this occurred because residual water in the solvent and M2 cooperatively coordinated with Mg2+. This allowed M2 to capture Mg2+ from MSP, which led to depolymerization. However, owing to the slow reaction between trace water/M2/Mg2+, the formation of MSP still occurred first. Therefore, water regulated both the thermodynamics and kinetics of the system and was the key factor for constructing the transient MSP. Fine-tuning the water content and other assembly motifs regulated the assembly evolution pathway, tuned the MSP lifetime, and made the luminescent color of the system undergo intriguing transition processes over time.
13 July 2022, PNAS
Shiyan Chen, Lixia Peng, Yanan Liu, Xiang Gao, Ying Zhang, Chun Tang, Zhenghao Zhai, Liulin Yang, Weitai Wu, Xumin He, Liu Leo Liu, Feng He and Haiping Xia*
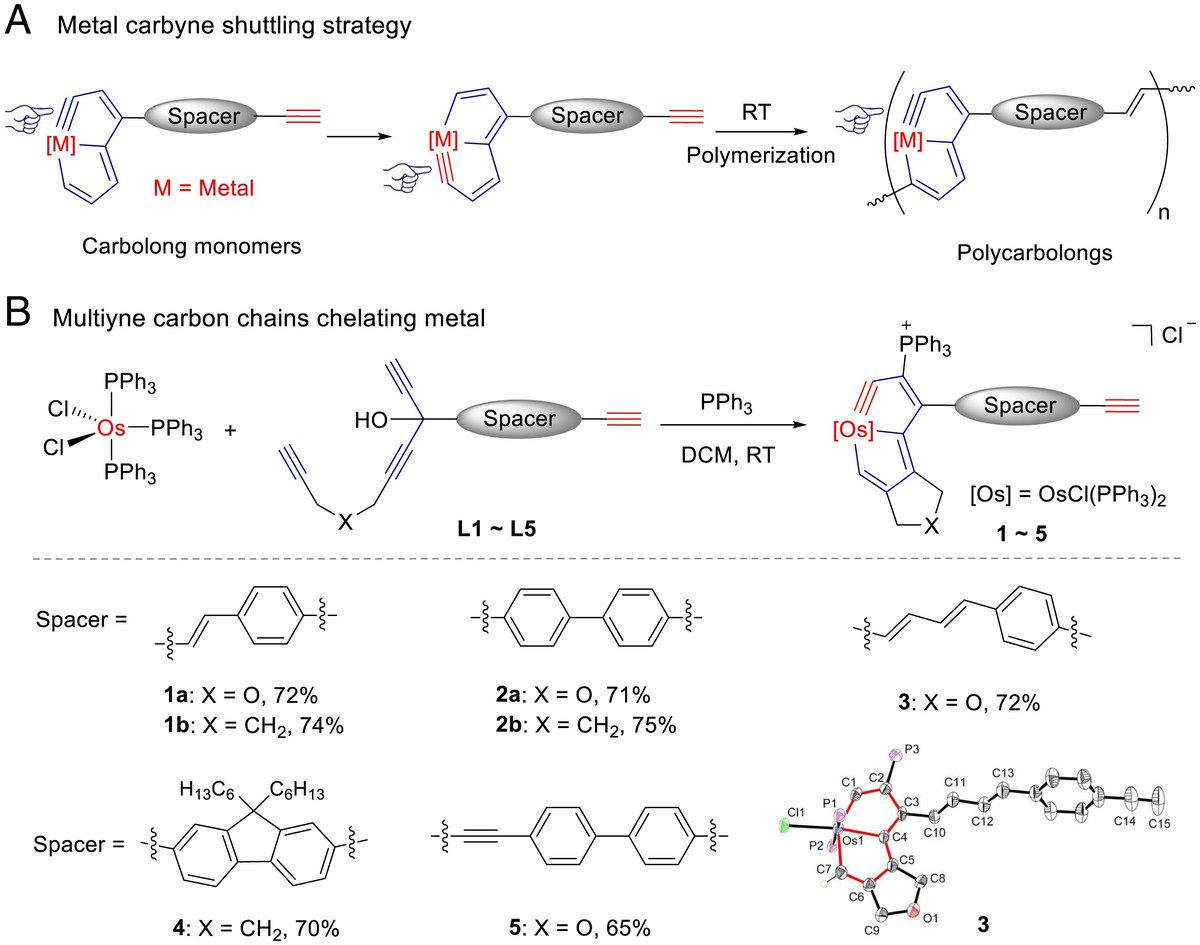
Conjugated polymers usually require strategies to expand the range of wavelengths absorbed and increase solubility. Developing effective strategies to enhance both properties remains challenging. Herein, we report syntheses of conjugated polymers based on a family of metalla-aromatic building blocks via a polymerization method involving consecutive carbyne shuttling processes. The involvement of metal d orbitals in aromatic systems efficiently reduces band gaps and enriches the electron transition pathways of the chromogenic repeat unit. These enable metalla-aromatic conjugated polymers to exhibit broad and strong ultraviolet–visible (UV–Vis) absorption bands. Bulky ligands on the metal suppress π–π stacking of polymer chains and thus increase solubility. These conjugated polymers show robust stability toward light, heat, water, and air. Kinetic studies using NMR experiments and UV–Vis spectroscopy, coupled with the isolation of well-defined model oligomers, revealed the polymerization mechanism.
17 Jun 2022, Chemical Science
Ganyu Chen, Peichen Shi, Longhui Zeng, Liubin Feng, Xiuxiu Wang, Xujing Lin, Yibin Sun, Hongxun Fang, Xiaoyu Cao, Xinchang Wang*, Liulin Yang* and Zhongqun Tian
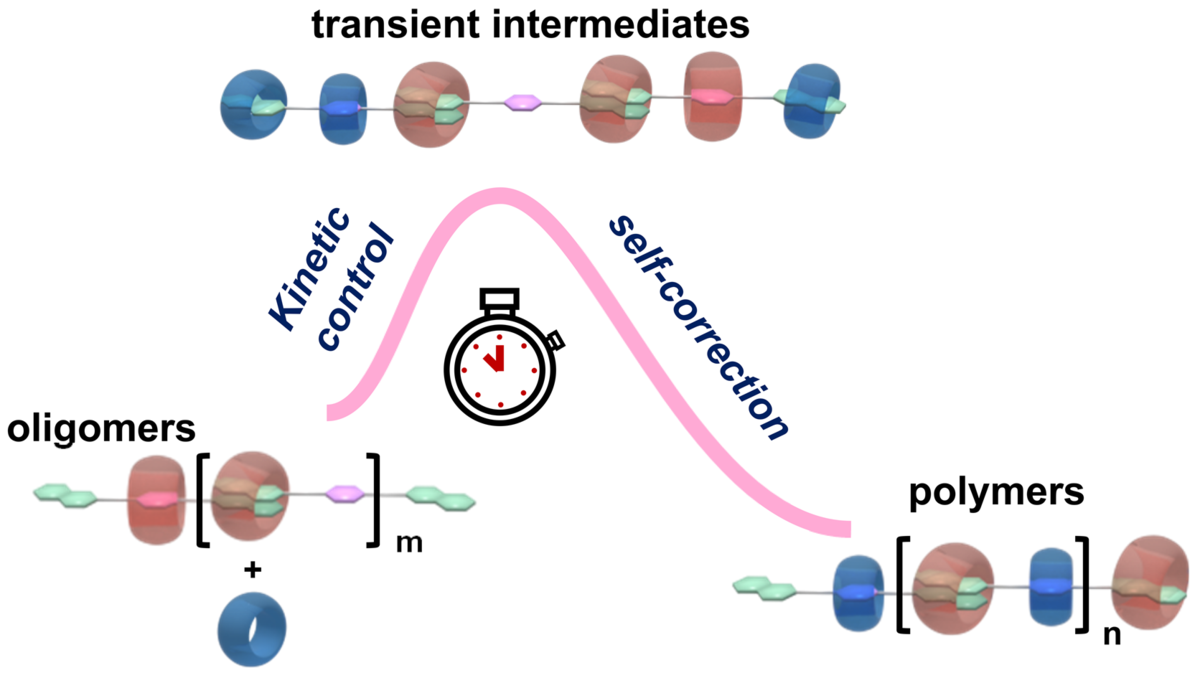
Kinetic control over structures and functions of complex assembly systems has aroused widespread interest. Understanding the complex pathway and transient intermediates is helpful to decipher how multiple components evolve into complex assemblies. However, for supramolecular polymerizations, thorough and quantitative kinetic analysis is often overlooked. Challenges remain in collecting the information of structure and content of transient intermediates in situ with high temporal and spatial resolution. Here, the unsolved evolution mechanism of a classical self-sorting supramolecular copolymerization system was addressed by employing multidimensional NMR techniques coupled with a microfluidic technique. Unexpected complex pathways were revealed and quantitatively analyzed. A counterintuitive pathway involving polymerization through the ‘error-correction’ of non-polymerizable transient intermediates was identified. Moreover, a ‘non-classical’ step-growth polymerization process controlled by the self-sorting mechanism was unraveled based on the kinetic study. Realizing the existence of transient intermediates during self-sorting can encourage the exploitation of this strategy to construct kinetic steady state assembly systems. Moreover, the strategy of coupling a microfluidic technique with various characterization techniques can provide a kinetic analysis toolkit for versatile assembly systems. The combined approach of coupling thermodynamic and kinetic analyses is indispensable for understanding the assembly mechanisms, the rules of emergence, and the engineering of complex assembly systems.
22 Apr 2022, Angew. Chem. Int. Ed.
Yang Chao, Dr. Tushar Ulhas Thikekar, Wangjian Fang, Rong Chang, Jiong Xu, Nianfeng Ouyang, Jun Xu, Yan Gao, Minjie Guo, Han Zuilhof, and Andrew C.-H. Sue*
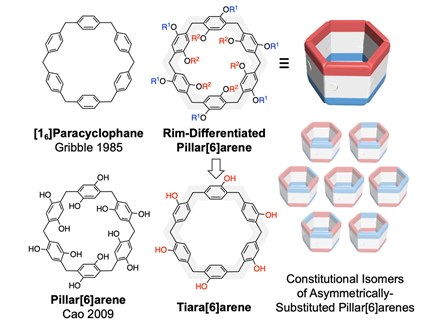
A “rim-differentiated” pillar[6]arene (RD-P[6]) was obtained successfully, with the assistance of a dimeric silver trifluoroacetate template, among eight different constitutional isomers in a direct and regioselective manner. The solid-state conformation of this macrocycle could switch from the 1,3,5-alternate to a truly rim-differentiated one upon guest inclusion. This highly symmetric RD-P[6] not only hosts metal-containing molecules inside its cavity, but also can form a pillar[6]arene-C60 adduct through co-crystallization on account of donor-acceptor interactions. The development of synthetic strategies to desymmetrize pillararenes offers new opportunities for engineering complex molecular architectures and organic electronic materials.
3 Mar 2022, Polymer Chemistry
Liulin Yang,‡ Haibo Zhao,‡ Yulin Xie, Pufan Ouyang, Yonghong Ruan, Jiangxi Chen, Wengui Weng, Xumin He*, Haiping Xia*
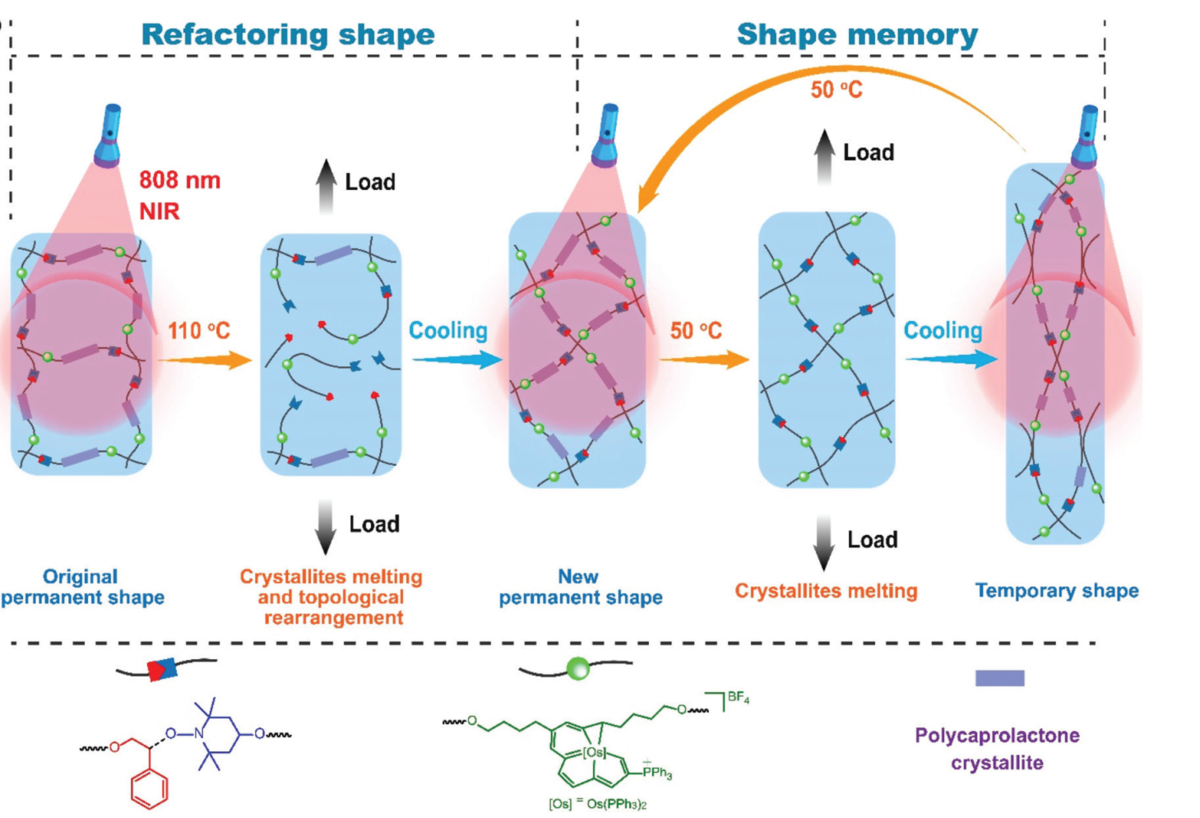
Conventional shape memory polymers (SMPs) are restricted to predetermined permanent shapes and therefore cannot be reconfigured arbitrarily to adapt to variant application scenarios. Meanwhile, shape memory behaviour is mostly thermally active and is often induced by direct heating and lacks spatial or remote control. Herein, we report a novel SMP with a reconfigurable network containing a semi-crystalline polymer chain, radically exchangeable covalent bond and photothermoresponsive carbolong complex moiety. The photothermal effect of the carbolong complex and the thermal responsiveness of the semi-crystalline polymer chain and radically exchangeable covalent bond lead to shape memory behaviour and network topological rearrangement using near-infrared light irradiation. Such a strategy offers an opportunity for building reconfigurable shape memory polymers that can be manipulated by either direct heating or remote light irradiation.
10 Feb 2022, Chem
Huihui Hu, Jieyu Zhu, Lingyun Cao, Zhiye Wang, Yuan Gao, Liulin Yang, Wenbin Lin, Cheng Wang*
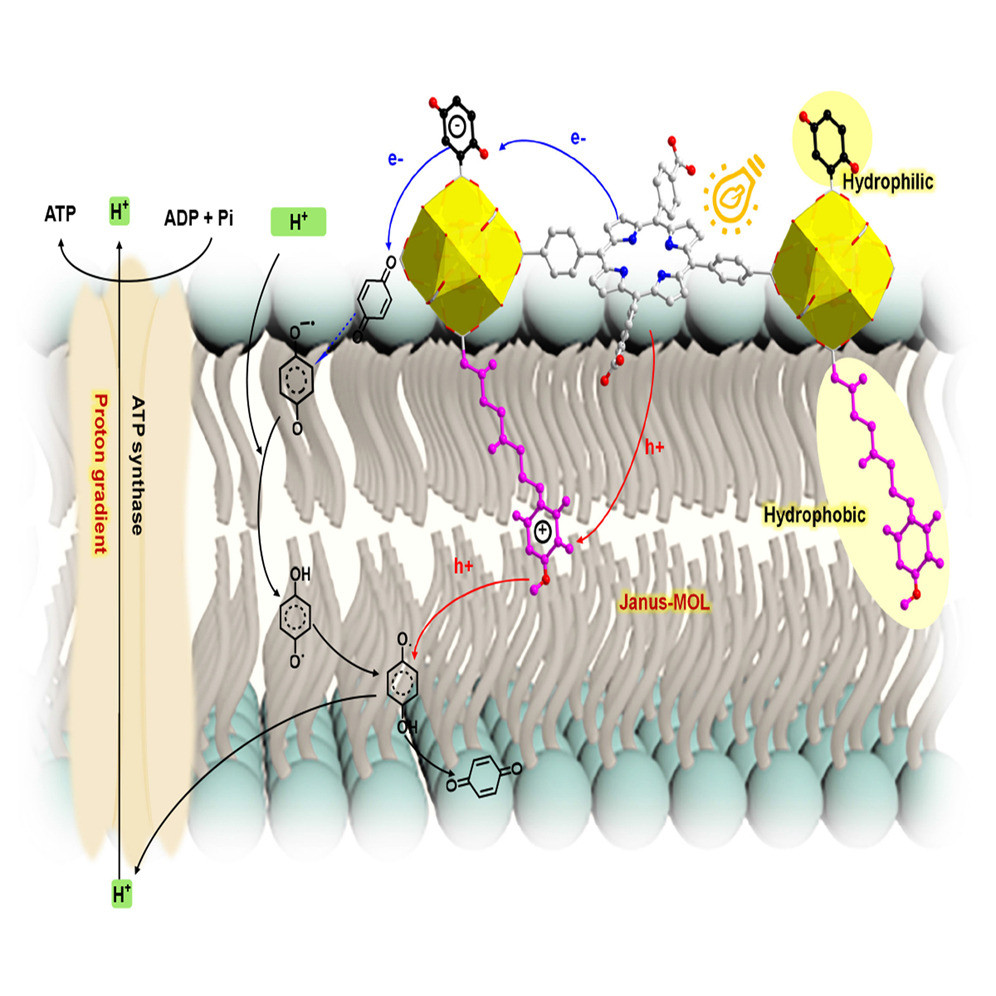
Photo-generation of a proton gradient over a lipid bilayer is of interest due to its essential role in photosynthetic bacteria. Membrane asymmetry is key to the proton gradient generation via directional proton transport. Here, we report a light-driven proton pump based on two-dimensional, porphyrin-based Janus metal-organic layers (Janus-MOLs). The Janus-MOL, functionalized with carboxyquinone on one side and Acitretin on the other via a microemulsion-based method, was attached to liposome surface. Upon photoexcitation, the porphyrins initiate electron and hole transfers to carboxyquinone and Acitretin, respectively, which undergo redox reactions with freely diffusing quinone (Q)/hydrosemiquinone (HQ·) in the lipid bilayer to produce a concentration gradient of quinone-based species. Owing to different pKa values of HQ+ and HQ·, these redox reactions trigger proton transport across the membrane to create a pH gradient, which drives ATP production by CFoF1-ATP synthase in a similar fashion as photosynthetic bacteria.







